A clinical trial is defined as the controlled, clinical testing in human subjects of investigational new drugs, devices, treatments or diagnostics, or comparisons of approved drugs, devices, treatments or diagnostics, to assess their safety, efficacy, benefits, costs, adverse reactions, and/or outcomes.
Such studies may be conducted under an industry-developed protocol (referred to as a "sponsor-initiated clinical trial") or an investigator-developed protocol (referred to as an "investigator-initiated clinical trial"). These studies are most often conducted in conjunction with obtaining new drug or device approval from the U.S. Food and Drug Administration (FDA), under an Investigational New Drug (IND) application or as IND exempt for drug studies (as *Phase I, II, III, or IV), or under an Investigational Device Exemption for device studies, although studies can be designed with the sole purpose of collecting and analyzing data about approved drugs or devices in order to contribute to medical knowledge about the treatment of a disease or medical conditions. In the case of a non-FDA regulated drug, device, treatment or diagnostic (including dietary supplements), the study must use medical facilities and University personnel in the performance of the study. In all cases, the study must include the prospective enrollment of human subjects and the controlled testing of a drug, device, treatment or diagnostic under an approved protocol.
To be eligible for using the University of California Office of the President (UCOP) approved Clinical Trial Indirect Cost Rate, financial support for a clinical trial must be provided by a for-profit, commercial entity, such as a pharmaceutical or device company. The above definition has no impact on the intellectual property policies applicable to various types of agreements, the allocation and distribution of private indirect cost recovery, applicability of various policies (including FDA regulations) about disclosure of conflict of interest, or other policies of the University.
Retrospective chart reviews, analysis of existing medical data and records, laboratory research, animal studies, and federally funded projects are not categorized as clinical trials for purposes of applying the UCOP approved clinical trial indirect cost rate.
Sponsored Programs Administration (SPA) is responsible for reviewing, negotiating and legally executing agreements from external funding sources.
Phase I:
The initial introduction of an investigational new drug into humans. They are typically closely monitored and may be conducted in patients or normal volunteer subjects. These studies are designed to determine the metabolism and pharmacologic actions of the drug in humans, and the side effects associated with increasing doses, and, if possible, to gain early evidence on effectiveness. During Phase I, sufficient information about the drug's pharmacokinetic and pharmacological effects should be obtained to permit the design of well-controlled, scientifically valid, Phase II studies. The total number of subjects and patients included in Phase I studies varies with the drug, but is generally in the range of 20 to 80. Phase I studies also include studies of drug metabolism, structure-activity relationships, and mechanism of action in humans, as well as studies in which investigational drugs are used as research tools to explore biological phenomena or disease processes.
Phase II:
Controlled clinical studies conducted to evaluate the effectiveness of the drug for a particular indication or indications in patients with the disease or condition under study and to determine the common short-term side effects and risks associated with the drug. Phase II studies are typically well-controlled, closely monitored, and conducted in a relatively small number of patients, usually involving no more than several hundred subjects.
Phase III:
Expanded controlled and uncontrolled trials. They are performed after preliminary evidence suggesting effectiveness of the drug has been obtained, and are intended to gather the additional information about effectiveness and safety needed to evaluate the overall benefit-risk relationship of the drug and to provide an adequate basis for physician labeling. Phase III studies usually include from several hundred to several thousand subjects.
Phase IV:
Conducted following a drug's approval from the FDA for a particular use and explore unapproved uses, dosages and indications of the drug.
-
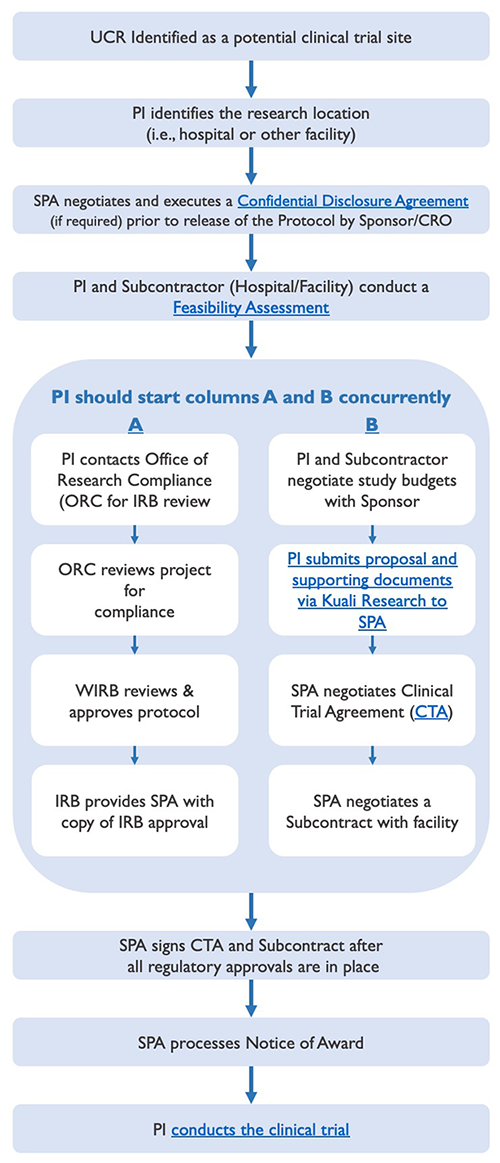
Lifecycle Process Leading to the Start of a Clinical Trial
Lifecycle Process Leading to the Start of a Clinical Trial
The lifecycle of pre-trial administration activities leading to the start of a clinical trial involves several key steps, some of which should occur simultaneously, as displayed below, to prevent undue delays.
Clinical Trial Lifecycle - (Phases I - IV)
-
Confidential Disclosure Agreements (CDA)
Confidential Disclosure Agreements (CDA)
A potential sponsor may want to send a protocol to the Principal Investigator (PI), so that he/she can decide whether to participate in the clinical trial. In the event the protocol contains information that the potential sponsor considers to be confidential or proprietary, it is not uncommon for such potential sponsor to request the execution of a confidential disclosure agreement ("CDA"), prior to the release of the protocol, as a means of protecting the disclosure of such information. (Note: CDAs are sometimes referred to as non-disclosure agreements ("NDAs").)
Should a PI receive a CDA requiring University signature, it will need to be reviewed, negotiated, and executed by Sponsored Programs Administration (SPA). CDA's are usually straightforward and require little negotiation; however, some CDA's can include requests that are contrary to UC policy and, thereby, require more time to resolve.
To process a CDA:
- Email the CDA to ClinicalTrials@ucr.edu (with a copy to your departmental analyst);
- Include the name, address, telephone number, and email address of the sponsor's (or that of any applicable contract research organization, aka "CRO") point of contact;
- Include the name, address, telephone number, and email address of the subcontractor's (i.e., the entity whose facility and/or clinical support services would be utilized by UCR in the conduct of the clinical trial) point of contract. Such potential subcontractor would also need to sign a CDA prior to receiving a copy of the protocol for review; and
- Include a note with any concerns about the CDA, if any.
Once the terms of the CDA have been negotiated, SPA and the sponsor (or CRO, if applicable) will execute the agreement. SPA will also coordinate the execution of the CDA for the entity that will be assisting in the feasibility assessment. (Note: The PI may be requested to sign an acknowledgement block indicating his/her willingness to comply with the terms of the CDA.) Thereafter, the fully executed CDA will be processed by SPA and a copy will be sent to the PI and his/her departmental analyst.
-
Feasibility Assessment
Feasibility Assessment
Upon receipt and subsequent review of a sponsor-developed protocol, the PI and applicable subcontractor (whose facility and/or clinical support services would be utilized by UCR in the conduct of the clinical trial) will jointly conduct a feasibility assessment to determine whether or not to move forward with the clinical trial. Such assessment should address the following questions:
- Does the protocol provide scientific value?
- Can study subjects be recruited, given the enrollment criteria?
- Does the budget support the work to be performed?
- Do we have the resources to conduct the research study?
Note: The determination to participate in the conduct of the clinical trial should be predicated on the ability to answer affirmatively to all of the above questions.
Additional factors to consider during the feasibility assessment process include Medicare coverage analysis, technical considerations, the particular services and facilities needed from the subcontractor for a given study, etc.
-
Submission Process - Initiating the Clinical Trial Submission Process
Initiating the Clinical Trial Submission Process
As soon as it appears likely that UCR will participate in a clinical trial, please process and route a proposal via the Proposal Development module in Kuali Research to Sponsored Programs Administration (SPA) with the following proposal documents uploaded as attachments:
- Clinical Study PI Questionnaire
- Request for Exception to Policy Regarding PI Eligibility Kuali Build form, if required
- Clinical Trial Protocol
- Draft Clinical Trial Agreement (CTA), if provided by sponsor (or contract research organization, referred to as "CRO")
- Draft CTA Study Budget
- Draft Subcontract Study Budget
Additionally, the following financial disclosures (as applicable) should be submitted to Research Compliance prior to routing of the proposal:
- Form 700-U (State of California's PI/co-PI Statement of Economic Interests), if the study sponsor is a non-governmental entity.
- Statement of Economic Interests Supplemental Information Form (submit this Addendum if the Form 700-U is positive)
- Federal Disclosure of Financial Interests Related to Sponsored Projects Form, if the study sponsor is a governmental entity (e.g., NIH funded study)
All of the above documents are required prior to the negotiation of the CTA with the study sponsor (or CRO). The review of the proposal and negotiation of the CTA requires time and, therefore, should not be left to the last minute. While SPA can proceed with the negotiation and execution of the CTA prior to IRB approval, the Notice of Award cannot be processed nor funds released until all regulatory approvals are in place (e.g., IRB approval of the protocol, informed consent, and HIPAA Authorization form). Therefore, the PI and study team are encouraged to proceed with other required pre-trial activities (e.g., finalizing the study budget, obtaining IRB approval, etc.) simultaneous to SPA's negotiation of the CTA to prevent undue delays.
-
Negotiation of Clinical Trial Agreements
Negotiation of Clinical Trial Agreements
Sponsored Programs Administration (SPA) is responsible for reviewing, negotiating, and executing agreements, including clinical trial agreements (CTAs), from external funding sources (e.g., pharmaceutical companies). As a public, nonprofit educational institution, the University is bound by certain policies and regulations regarding what it can and cannot accept in a CTA. These policies are designed to protect the welfare of individuals participating as a research subject; foster the University's basic mission of teaching, research, and public service; and minimize the various forms of liability associated with human research.
Principal Investigators (PIs) are encouraged to discuss all aspects of the clinical trial with SPA prior to the start of negotiations. For instance, apprising SPA of the PI's involvement in the protocol development and/or study design raises intellectual property implications that may result in the negotiation of different or additional contractual terms.
The contractual language proposed by the University during contract negotiations is predicated upon the following:
- That the clinical investigation is conducted under a protocol that is a FDA Phase I, II, II, or IV drug study or a FDA regulated medical device study;
- That the sponsor provides its proprietary product (e.g., drug and/or device) to the University for the purpose of conducting the sponsor-initiated or investigator-initiated clinical trial;
- That the sponsor provides its study protocol to the University, if it is a sponsor-initiated clinical trial
- That the PI is the author of the study protocol, if it is an investigator-initiated clinical trial; and
- That the sponsor will fully fund the cost of the study (i.e., no work will be supported in whole or in part with other funds),
When negotiating CTAs, the University's primary focus is on securing acceptable contract clauses regarding high-risk issues (e.g., indemnification, confidentiality, publication rights, data ownership, patent rights, and subject injury).
Although each agreement is reviewed on a case-by-case basis, there are a number of key contractual provisions that are common to most CTAs. Some of those provisions and the accompanying explanation as to the University's handling thereof are as follows:
Parties to the Agreement
CTAs should only be made by and between the study sponsor and The Regents of the University of California, on behalf of its Riverside campus. The PI, although an employee of UCR, does not have the delegated legal authority to enter into CTAs on behalf of The Regents and, therefore, may not be a named party to the agreement. That said, an acknowledgement block may be included at the end of the CTA whereby the PI may acknowledge that s/he has read the terms of the CTA and understands his/her obligations to comply therewith.
Note: Since UCR does not own a medical facility conducive to conducting clinical trials, UCR will need to contract with another entity for the use of their clinical facility and/or clinical support services. While such contractual arrangement will typically be handled in a separate agreement (e.g., subcontract) between The Regents and that other entity, there could be instances where a study sponsor, instead, may require such other party to be a named party to the CTA.
Financial Arrangements
CTA typically contain a payment schedule which includes a per subject fixed-price. Such schedules usually provide for periodic payments at regular intervals or payments upon the completion of milestones (e.g., enrollment of a certain percentage of subjects, submission of a certain number of case report forms, etc.).
The University cannot underwrite expenses for the sponsor; therefore, the University requires a minimum advance payment, typically between of 10-20% of the total anticipated cost, upon execution of the agreement. When a project involves significant start-up costs, the University may require a larger initial payment.
Indemnification
When conducting a sponsor-initiated clinical trial whereby the sponsor is the author of the protocol, the University is following the sponsor's instructions. As a public, non-profit educational institution, UCR cannot bear the financial responsibility for any injury (including damages) resulting from the performance of such a clinical trial. Consequently, UCR requires the sponsor to maintain a policy or program of insurance sufficient to support its indemnification obligation. The sponsor's obligation to assume all financial responsibility, however, does not apply to injury that is: (i) caused by UCR's failure to adhere to the protocol; (ii) failure to comply with FDA or other governmental requirements; or (iii) caused by the negligence of a faculty member or the University.
Confidentiality
UCR must maintain an open academic environment to fulfill its mission, and meet its fiduciary responsibilities, as a public educational institution. Clinical trials involving the use of a sponsor's confidential information should be limited to that information necessary for the performance of the clinical trial, and clearly identified by the sponsor as confidential.
Raw source data or documentation generated by the University during the conduct of a clinical study cannot be considered to be owned by the sponsor, nor can it be considered or treated as confidential information.
In addition, any agreements entered into by the University are subject to public disclosure under the State of California Public Records Act.
Publication
Timely publication and dissemination of research/study results are important principles behind the academic freedom afforded to each UCR faculty member. The publication provision, however, may provide a sponsor with a pre-publication review and comment period during which the sponsor may request the removal of its confidential information from the proposed publication. Additionally, the sponsor may promptly make arrangements for the protection of its intellectual property prior to publication. UCR is also willing to delay dissemination of study results for a reasonable period of time to accommodate multi-site studies. However, the resulting agreement cannot restrict UCR faculty from freely publishing UCR's research/study results.
Intellectual Property
Sponsors of UCR research are usually granted patent rights in accordance with University policies. However, UCR may grant greater rights to sponsors of sponsor-initiated clinical trials that meet all of the following criteria:
- The clinical investigation is an FDA Phase I, II, III or IV drug study or an FDA regulated medical device study.
- The sponsor provides its proprietary product and study protocol to the University for the clinical investigation.
- The cost of the clinical investigation, conducted according to the sponsor's protocol, is fully funded by the sponsor and is not supported, in whole or in part, with any other funds, including Federal funds, gift funds or foundation funds.
- There are no known third-party rights to intellectual property of The Regents that would be compromised by granting rights to the clinical trial sponsor.
- All University requirements regarding the administration of agreements with private sponsors for drug and device testing using human subjects have been satisfied.
If all of the above criteria are met, UCR may grant to the sponsor a certain range of rights to inventions made in the direct performance of the clinical trial protocol; provided, however, that with regard to Phase III or IV trials, there must have been little or no investigator involvement in the conception or development of the protocol. Notwithstanding the foregoing, UCR reviews patent terms on a case-by-case basis and prefers to do so with a thorough understanding of the work contemplated.
Subject Injury
As a matter of UC policy, in the event a study subject is injured resulting directly from the administration of study drug or the study procedures carried out in accordance with a sponsor-designed protocol, UCR will provide reasonably necessary medical treatment. UC policy specifically prohibits billing the study subject or a third-party for the costs of treating such injuries. Therefore, UCR requires reimbursement for the cost of such treatment from the sponsor. Human subject welfare is a primary concern for the UCR and exceptions to these terms cannot be accommodated.
Insurance
To support its indemnification obligations, the sponsor must maintain a sufficient level of insurance.
The University of California is self-insured and, during the term of the agreement, will maintain in force adequate insurance to cover its indemnification obligations.
Governing Law
The University of California is a constitutional corporation of the State of California and contracts accepted by the University are to be interpreted under California Law. UCR will also consider contractual silence regarding this issue.
Termination
Each of the parties to the CTA must have the ability to terminate, upon the giving of reasonable notice. In the event of early termination, UCR will seek reimbursement from the sponsor for all costs and uncancellable obligations incurred up to the date of termination. In addition, untimely withdrawal from the protocol could jeopardize the welfare of human subjects; therefore, the UCR shall request that the sponsor cooperate with UCR to safely withdraw subjects from the protocol.
Use of Name
California Education Code Section 92000 provides that the use of the name "University of California" is the property of the State of California and that the sponsor's use of such name, campus, or logo must also comply with such section. Accordingly, the sponsor is prohibited from using UCR's name in connection with any advertisement, press release, or other form of business promotion or publicly, or to suggest that it endorses a product or service. A sponsor may, however, use UCR's name to fulfill its obligations as required by law, or by otherwise requesting prior written approval from UCR.
-
Clinical Trial Budget Considerations
Clinical Trial Budget Considerations
Generally, industry sponsors are concerned with the total cost of conducting a project rather than the classification of costs. As a consequence, the structures of most clinical trial budgets differ from those of grant proposal budgets. Sponsor-provided budget templates are typically separated into up-front costs (e.g., advertising costs associated with patient recruitment, pharmacy set-up, initial IRB review fee, translation service for informed consent, etc.), per patient costs, and items to be invoiced on an as-needed basis (e.g., screen failures). Other budget issues for consideration are listed below:
Departmental Overhead
When preparing and negotiating the budget for an industry sponsored clinical trial, be sure to cover all costs associated with conducting the trial. Due to the unique administrative nature of clinical trials and the reduced facilities and administrative rate currently being applied to clinical trials, charging administrative expenses may be appropriate if the sponsor will cover these costs as a direct charge.
Patient Care Costs and Coverage Analysis
To obtain a coverage analysis and current research rates for patient care costs, including Pharmacy services, contact paul.hackman@medsch.ucr.edu.
IRB Fees
The Institutional Review Board (IRB) charges fees for the initial review of clinical trials from for-profit sponsors as well as a subsequent IRB renewal fee should the clinical trial extend beyond the date of the initial IRB approval.
Travel
Costs for travel to Investigator's Meetings or other study-related meetings may be managed in a variety of ways. Be sure to clarify how such costs should be incorporated into your study budget, if appropriate. Generally, pharmaceutical sponsors of clinical trials will not allow the budgeting of travel. Some pharmaceutical sponsors may allow travel costs associated with UCR employees traveling to different local area performance sites for conducting the clinical trial. In addition, some sponsors opt to directly reimburse UCR investigators for travel to meetings associated with conducting the clinical trial for the sponsor.
When using a sponsor's budget form, if travel is anticipated but the form lacks a line item or category for travel, complete the budget form and include travel costs as appropriate for each budget item. If investigator travel will be reimbursed by the sponsor, notify SPA, so that the CTA will reflect this understanding.
Facilities & Administration Fees
As a public-supported institution, UCR must recover the full cost of research conducted for outside sponsors, including all associated operating costs ("indirect" costs). To do otherwise could result in subsidizing for-profit research with public funds. Indirect costs are facilities and administrative ("F&A") costs incurred in support of the University's research infrastructure. The University pools its indirect costs for ease of accounting because it is difficult to assign these costs with a relative degree of accuracy to a specific project or program. The federal government's Office of Management and Budget establishes the standards for calculating the F&A cost rate, and UCR negotiates its rates with the United States Department of Health and Human Services audit agency on a periodic basis. UCR derives its F&A rate for clinical studies from applicable components of the federally approved rate.
As an exception to recovery of our federally-negotiated Facilities & Administrative cost rate applicable to research at UCR, costs applicable to research at UCR which meets the definition of a clinical trial (i.e., "the controlled, clinical testing in human subjects of investigational new drugs, devices, treatments or diagnostics, or comparisons of approved drugs, devices, treatments or diagnostics, to assess their safety, efficacy, benefits, costs, adverse reactions, and/or outcomes"), and for which is not funded with federal funds, are subject to a 26% indirect cost rate. Such 26% indirect cost rate applies to the Total Direct Cost, and no budgeted item is excluded from the application of indirect costs. This rate applies regardless of whether a clinical trial is based on a sponsor-initiated or an investigator-Initiated protocol.
Below are FAQs to ponder when determining whether or not the clinical trial rate of 26% TDC is appropriate for a given study.
FAQ - Application of F&A Cost Rates for Clinical Trials
A study meeting the UCR definition of a clinical trial will be conducted as a cooperative group trial under NIH funding. Can the clinical trial facilities and administrative cost rate of 26% TDC be used?
No. Studies supported in whole or in part with federal funding must apply the applicable Federal research facilities and administrative cost rate.
A study will be funded by an industry sponsor via a subcontract to UCR, with prime funding provided by the Federal government. Since the funds will be received directly from the industry sponsor, can the clinical trial indirect cost rate of 26% TDC be applied?
No, because Federal flow-through funds are involved; the applicable Federal research F&A cost rate should be applied.
A study fully funded by a for-profit entity will involve testing of an investigational drug or device in animals. Which facilities and administrative cost rate applies?
The clinical trial indirect rate only applies to clinical testing in human subjects. Pre-clinical laboratory and animal studies are specifically excluded. Therefore, the applicable Federally-negotiated research F&A cost rate would apply.
A pharmaceutical company has requested that UCR run specialized tests on samples obtained in a clinical trial. Would the clinical trial indirect cost rate apply?
No. The clinical trial indirect cost rate only applies to studies involving the direct clinical testing of investigational products in human subjects. If UCR's sole involvement is to run tests, the applicable Other Sponsored Activities rate would apply.
An investigator has agreed to analyze data collected in an industry-sponsored clinical trial conducted at both UCR and other sites. Which facilities and administrative cost rate applies?
The applicable Federally-negotiated research rate would apply.
The University has been asked by a for-profit sponsor to recruit and enroll human subjects in order to collect data that will be submitted to a patient registry. Is this considered a clinical trial, and would the clinical trial indirect cost rate apply?
Data collection studies for registry purposes, even if human subjects are involved, are not considered clinical trials. Therefore, the clinical trial indirect cost rate would not apply, and the applicable Other Sponsored Activities facilities and administrative cost rate should be used.
-
Conducting the Clinical Trial
Conducting the Clinical Trial
Launching of the Clinical Trial
The clinical trial cannot commence until all regulatory compliance approvals are in place (e.g., IRB approval of the protocol, informed consent, and HIPAA Authorization form) and, thereafter, SPA has: (i) signed and is in receipt of a fully executed CTA and (ii) fully executed the clinical trial subcontract with the facility whose premise and/or clinical support services will be utilized. (Again, SPA may negotiate the CTA simultaneously with the IRB process; it is just that the contractual signature process must be delayed until all regulatory approvals are in place. Thereafter, the performance of the clinical trial may begin.)
Clinical Trial Site Visits
The ability of a sponsor of a clinical trial to conduct a site visit depends upon who authored the protocol (i.e., the sponsor or the Principal Investigator (PI)). A clinical trial where the protocol is written by the sponsor is referred to as a sponsor-initiated clinical trial. A clinical trial where the PI is the author of the protocol is referred to as an investigator-initiated clinical trial.
Sponsor-Initiated Clinical Trials
Under sponsor-initiated clinical trials, the sponsor, or its authorized representative, will be entitled to examine, at mutually agreeable times during normal business hours, the facilities where the clinical trial is being conducted, the trial data, and other relevant information necessary to confirm that the trial is being conducted in accordance with the terms of the CTA, the protocol, and applicable laws and FDA regulations. A sponsor, or its representative, that is provided access to source records for the purpose of monitoring or auditing the clinical trial cannot make a record of or disclose direct identifiers of any participating patient in the trial (including the patient's name, street address, telephone, social security, medical record, or health plan beneficiary numbers). In the event that copies of the original source records are to be made, all direct identifiers of patients participating in the clinical trial must be redacted prior to copying or taking copies of the source documents off site.
Investigator-Initiated Clinical Trials
Under investigator-initiated clinical trials, the Institution and PI are solely responsible for the monitoring of the clinical trial in compliance with good clinical practices. Visits by the sponsor shall be limited to the review of clinical data for the sole purpose of pursuing regulatory filings related to the study drug or device. The review of such data cannot contain patient identifiers.